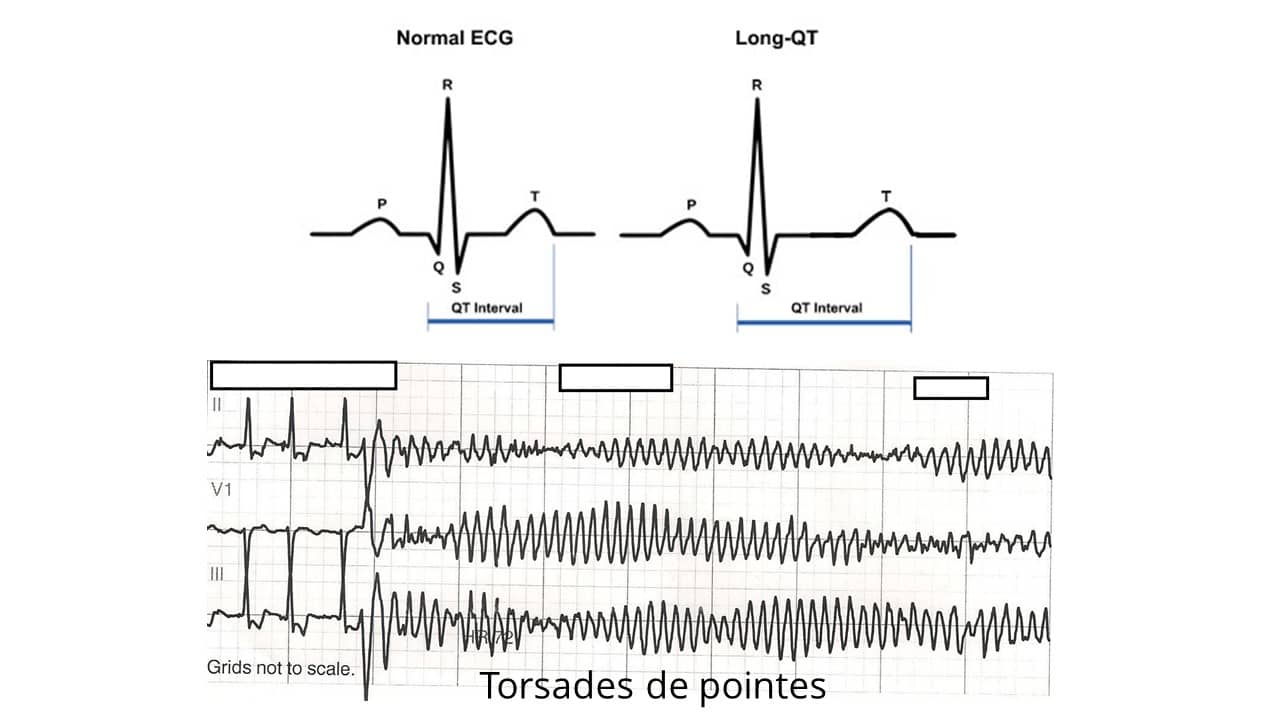
A síndrome do QT longo é uma entidade importante, sendo o seu reconhecimento fundamental para a tomada de decisão clínica. A forma adquirida está associada ao uso de drogas ou a certas condições clínicas comumente encontradas na prática médica diária, como por exemplo, distúrbios eletrolíticos como a hipopotassemia, a hipomagnesemia e a hipocalcemia. A sua forma congênita é causa de síncope e morte súbita em jovens e crianças. Como os testes genéticos ainda não são de uso corrente, o diagnóstico na prática é feito com base em dados clínico-eletrocardiográficos. O intervalo QT deve sempre ser corrigido para a frequência cardíaca, porém é importante notar que não somente a duração do QT, mas também alterações morfológicas da onda T são importantes para o diagnóstico. Identificar alterações patológicas do intervalo QT é fundamental para evitar arritmias ventriculares malignas, que estão associadas a esta condição.
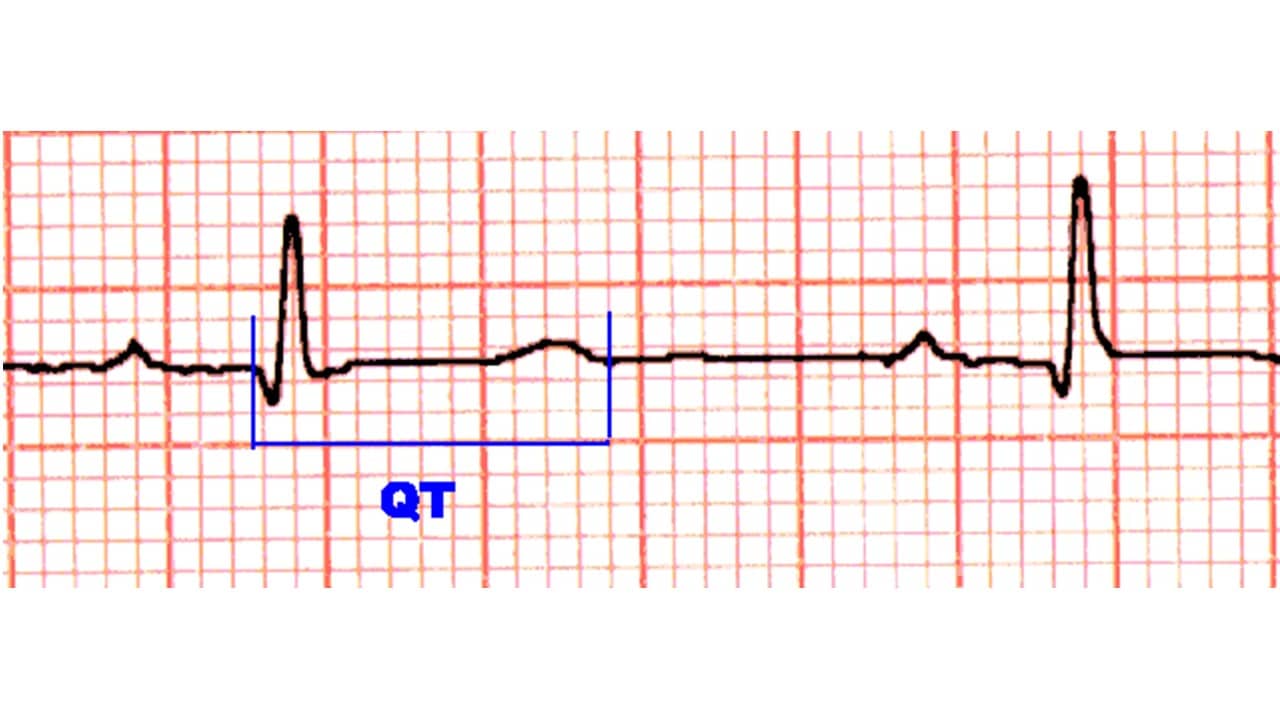
O intervalo QT é definido como a medida do início do complexo QRS até o final da onda T. Este intervalo representa a duração total da atividade elétrica ventricular. A fórmula de Bazzet (abaixo) é amplamente utilizada para cálculo do QT corrigido, mas apresenta limitações para frequência cardíaca < 60 ou > 90 bpm. Dessa forma, a diretriz brasileira para emissão de laudos eletrocardiográficos recomenda utilizar fórmulas lineares como a de Framingham e a de Hodges (atualmente disponível em aplicativos para smartphones). O valor normal do intervalo QT é menor ou igual a 450 para homens e 470 ms para mulheres. Para crianças o valor de referência é de até 460 ms. O aumento do intervalo QT pode ser devido tanto a agentes externos quanto a alterações geneticamente determinadas por mutações em canais iônicos cardíacos. Chamamos respectivamente de adquirido ou congênito a estes dois tipos de síndrome do QT longo.
Fórmula de Bazzet:
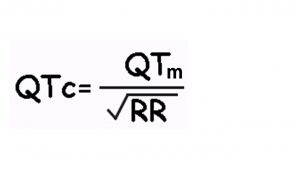
QTc= Intervalo QT corrigido pela freqüência cardíaca.
QTm= Intervalo QT medido no ECG.
RR= Intervalo RR medido no ECG.
A arritmia ventricular associada ao aumento do intervalo QT é um tipo de taquicardia ventricular polimórfica distinta chamada de Torsades de pointes (torsão ou rotação das pontas), devido ao seu aspecto sinusoidal (Figura abaixo).
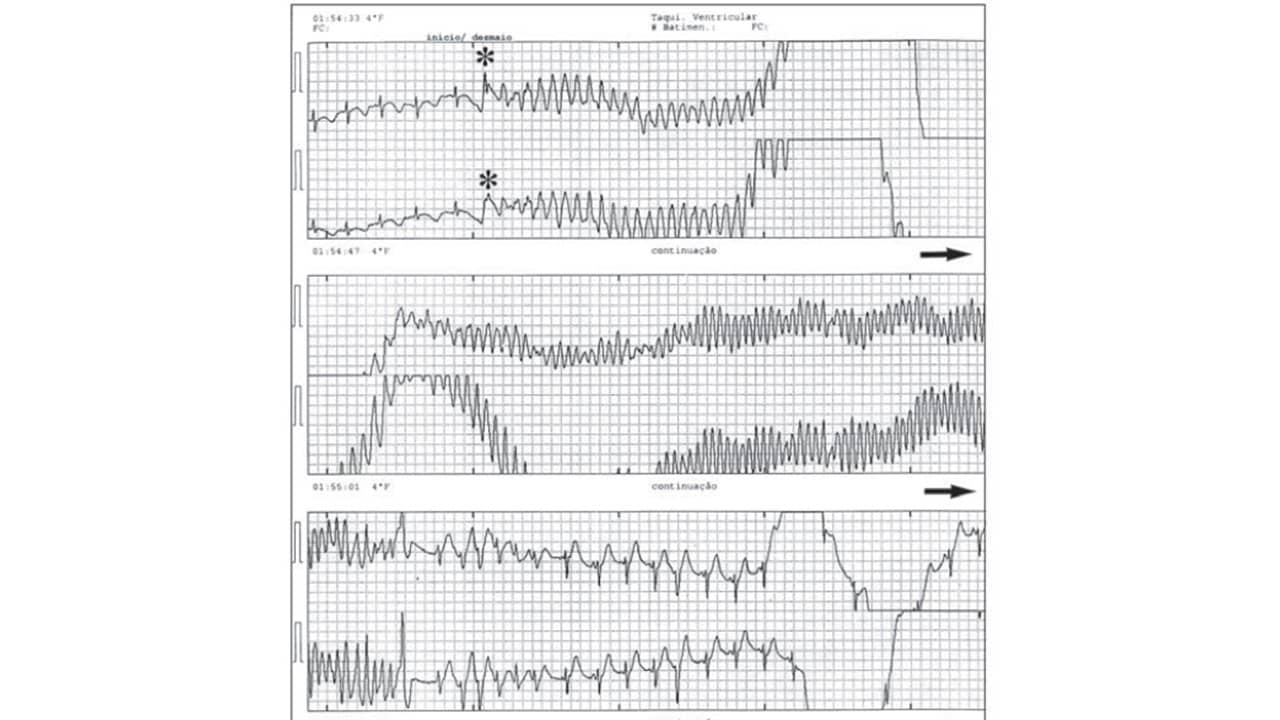
Vários trabalhos experimentais e modelos matemáticos demonstraram que o mecanismo indutor desta arritmia seria uma extrassístole devido à despolarização diastólica precoce ou à atividade “trigada”, e o perpetuador seriam reentradas nas áreas de não-homogeneidade de repolarização. Classicamente o torsades de pointes está associado à presença de uma sequência de ciclo longo-ciclo curto precedendo sua indução, porém mais recentemente demonstrou-se que o maior risco de indução é associado a uma modesta aceleração da frequência cardíaca, partindo de um estado inicial de bradicardia. O torsades de pointes pode exibir degeneração em fibrilação ventricular, sendo o mecanismo arrítmico de morte súbita em pacientes com QT longo.
A Síndrome do QT Longo Adquirida:
Inúmeras medicações e algumas condições metabólicas podem promover aumento do intervalo QT. No quadro abaixo são mostradas as principais drogas e condições eletrolítcas que estão fortemente implicadas no aparecimento de QT longo e torsade de pointes. Uma lista completa pode ser obtida em https://crediblemeds.org/healthcare-providers/ . Em adultos com QT corrigido alargado, devemos sempre pensar em causas externas. Como sempre, nós editores do CardioSite criamos um método mnemônico para nossos alunos lembrarem facilmente dessas condições que podem alargar o intervalo QT. Nosso método é o ABCDEFHM conforme tabela abaixo.
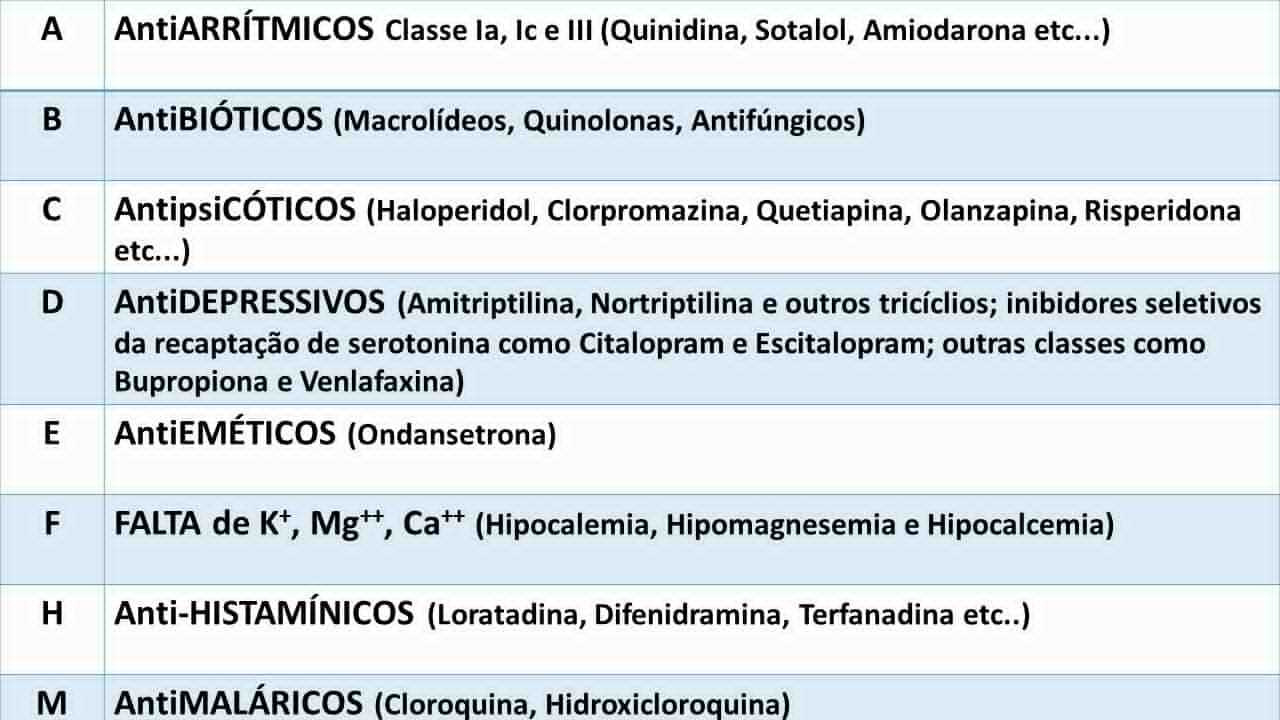
Conclusão:
A síndrome do QT longo apresenta um espectro muito grande de alterações, incluindo desde mutações genéticas até interações farmacológicas complexas e doenças que cursam com distúrbios eletrolíticos, por vezes de difícil diagnóstico. O médico deve estar atento aos sinais eletrocardiográficos únicos desta entidade, que por vezes podem ser a pista fundamental para o diagnóstico e tratamento correto desses pacientes.






